Laura M. Bohn, Agnes Acevedo-Canabal, Nicole Kennedy, Thomas Bannister, Edward L Stahl.
UF Scripps Biomedical Research, Jupiter FL 33458 USA
Biased agonism at the mu opioid receptor has been sought to improve opioid-mediated pain relief while avoiding adverse side effects. In particular, agonists that show a relative improvement in their ability to promote G protein signaling over βarrestin2 recruitment to MOR have been sought, and reported by many different groups. We have developed a class of these agonists that display a wide spectrum of bias; some agonists impart preferential signaling to G proteins, while some induce preferential recruitment of βarrestin2. Recently, we reported that some of the G protein signaling biased agonists are also tight binding, non-competitive agonists. This class of compounds, having wide ranging pharmacological properties, including partial and full efficacy agonists, will help us understand which properties may be best predictive of improved therapeutic outcomes. Moreover, they will help us understand, from a mechanistic perspective, of what causes the receptor to appear to prefer certain interactions over others.
Funding is from NIDA (5R01DA033073, R01DA038964, F32DA052124 (Agnes Acevedo-Canabal).
Yan Zhang
Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University
Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University
Opioid use disorders (OUD) pose an imminent threat to human health worldwide with approximately 2.1 million Americans suffering from this epidemic. Along with the analgesia produced by opioids, their ability to cause euphoria, due to activation of the mu-opioid receptor (MOR) in several regions of the brain, often leads to opioid misuse. Currently, detoxification and maintenance therapy are the two mostly used approaches to treat OUD. Methadone, buprenorphine and naltrexone (NTX) are first-line opioid medicines approved by the US Food and Drug Administration (FDA) for OUD. However, they carry some concerning side effects like the withdrawal symptoms precipitated by NTX and naloxone (NLX), including abdominal cramps, nausea/vomiting, diarrhea, muscle aches, anxiety, confusion, and extreme sleepiness. High doses of these drugs are also reported to show hepatotoxicity, cardiovascular and pulmonary problems. Thus, there is an urgent need to develop highly potent, efficacious, and selective MOR ligands with minimum side effects as OUD medications. In our lab, NAQ was identified as a selective, high potency and low efficacy MOR partial agonist. NAQ carries acceptable ADMET properties and improved oral bioavailability over naloxone (NLX) and naltrexone (NTX). At the in vivo level, NAQ displayed potent inhibition of the analgesic effect of morphine while precipitating fewer withdrawal symptoms in morphine dependent mice than NLX and NTX. In self administration studies, NAQ (10 and 32 mg/kg/day) produced behavioral reallocation away from heroin and towards the alternative nondrug food reinforcer. Overall, NAQ seems a promising molecule for developing novel opioid abuse and addiction therapeutic agents. Future studies including formulation, oral effective dose for OUD treatment, in vivo metabolism profiling, acute and chronic toxicities are warranted.
Andrew S. Huhn
Psychiatry and Behavioral Sciences – Behavioral Biology Research Unit, Johns Hopkins University
Background: There is an urgent need to translate preclinical findings into pharmacotherapeutic strategies for persons with opioid use disorder (OUD). There is strong preclinical evidence that orexin receptor antagonists can improve sleep, opioid withdrawal, and drug seeking outcomes, yet the effects of orexin receptor antagonists have not been tested in humans with OUD. This randomized-controlled trial aimed to examine whether a dual orexin receptor antagonist, suvorexant, would improve sleep duration, opioid withdrawal, and craving outcomes in persons undergoing a buprenorphine taper.
Methods: Persons with OUD were enrolled in a 10-night residential study (clinical trials registration: NCT03789214). All participants were initially inducted and maintained on 8-16mg buprenorphine for three days, then randomized to either placebo, 20mg, or 40mg SUVO before starting a 4-day buprenorphine taper and 4-day post-taper period. Each night participants wore a 3-lead electroencephalography (EEG) device to monitor sleep, and completed daily ratings of opioid withdrawal (Subjective Opioid Withdrawal Scale; SOWS), and 0-100 visual analogue scale (VAS) “Craving”. Chi-square analysis was utilized to assess differences in treatment attrition, intent-to-treat analyses were performed using independent-samples t-tests for differences between suvorexant (collapsed across dose) and placebo during the taper and post-taper period, and one-way ANOVAs were used to assess differences between all three medication groups.
Results: Participants (N=38) were 86.8% male, 52.6% white, and had a mean (SD) age of 41.5 (11.3). Participant attrition was not significantly different between placebo (33.3%), 20mg (n=35.7%), and 40mg (n=25.0%) groups. During the buprenorphine taper, participants on suvorexant versus placebo displayed a mean (SE) improvement of 91.2 (36.5) minutes in TST (p=.010), a marginal reduction of 2.3 (2.4) in mean (SE) SOWS (p=.325), and a mean (SE) reduction of 16.7 (8.1) in VAS craving (p=.046). During the post-taper period, participants on suvorexant versus placebo displayed a mean (SE) improvement of 61.2 (30.3) minutes in TST (p=.033), a mean (SE) reduction of 4.5 (2.1) in SOWS (p=.038), and a marginal mean (SE) reduction of 14.6 (9.3) in VAS craving (p=.13). ANOVAs revealed no differences between the 20mg and 40mg doses of suvorexant.
Conclusions: This preliminary, dose-finding study demonstrated initial efficacy for suvorexant in improving sleep duration, decreasing craving during opioid tapering, and improving post-taper withdrawal severity. Given the lack of suvorexant dose effects, future studies are needed to examine the FDA-approved dose of 20mg SUVO to confirm its efficacy in treating sleep disturbances, opioid withdrawal, and craving in persons with OUD.
Methods: Persons with OUD were enrolled in a 10-night residential study (clinical trials registration: NCT03789214). All participants were initially inducted and maintained on 8-16mg buprenorphine for three days, then randomized to either placebo, 20mg, or 40mg SUVO before starting a 4-day buprenorphine taper and 4-day post-taper period. Each night participants wore a 3-lead electroencephalography (EEG) device to monitor sleep, and completed daily ratings of opioid withdrawal (Subjective Opioid Withdrawal Scale; SOWS), and 0-100 visual analogue scale (VAS) “Craving”. Chi-square analysis was utilized to assess differences in treatment attrition, intent-to-treat analyses were performed using independent-samples t-tests for differences between suvorexant (collapsed across dose) and placebo during the taper and post-taper period, and one-way ANOVAs were used to assess differences between all three medication groups.
Results: Participants (N=38) were 86.8% male, 52.6% white, and had a mean (SD) age of 41.5 (11.3). Participant attrition was not significantly different between placebo (33.3%), 20mg (n=35.7%), and 40mg (n=25.0%) groups. During the buprenorphine taper, participants on suvorexant versus placebo displayed a mean (SE) improvement of 91.2 (36.5) minutes in TST (p=.010), a marginal reduction of 2.3 (2.4) in mean (SE) SOWS (p=.325), and a mean (SE) reduction of 16.7 (8.1) in VAS craving (p=.046). During the post-taper period, participants on suvorexant versus placebo displayed a mean (SE) improvement of 61.2 (30.3) minutes in TST (p=.033), a mean (SE) reduction of 4.5 (2.1) in SOWS (p=.038), and a marginal mean (SE) reduction of 14.6 (9.3) in VAS craving (p=.13). ANOVAs revealed no differences between the 20mg and 40mg doses of suvorexant.
Conclusions: This preliminary, dose-finding study demonstrated initial efficacy for suvorexant in improving sleep duration, decreasing craving during opioid tapering, and improving post-taper withdrawal severity. Given the lack of suvorexant dose effects, future studies are needed to examine the FDA-approved dose of 20mg SUVO to confirm its efficacy in treating sleep disturbances, opioid withdrawal, and craving in persons with OUD.
Yingpeng Liu, Lipin Ji, Fei Tong, Markos Orestis Georgiadis, Spyros P. Nikas, Alexandros Makriyannis.
Center for Drug Discovery, Northeastern University
Center for Drug Discovery, Northeastern University
Our knowledge of the chemistry and biology of the endocannabinoids anandamide (AEA) and 2-AG is rapidly expanding with the discoveries of new biological roles, novel pharmacological targets and signaling pathways as well as enzymes responsible for their synthesis, hydrolysis, transport, and oxidative metabolism of these lipid mediators. As such, to explore the biological functions and pharmacology of the endocannabinoid system, metabolically stable analogs of AEA and 2-AG are needed. Incorporation of structural modifications to enhance the biological activities and target specificities of these lipids while increasing their metabolic stabilities, is a challenge. In our approach we explore two design principles: a) the introduction of chiral methyl groups and polar moieties at judiciously chosen positions within the arachidonoyl chain of the prototype, and b) the inclusion of steric bulk and stereochemistry in the vicinity of the hydrolysable head group of the lipid. Herein we report novel arachidonoyl ethanolamide (AEA) and 2-arachidonoyl glycerol (2-AG) analogs that combine distinct conformational properties with unique functional profiles at the cannabinoid CB1 and CB2 receptors, coupled with enhanced stabilities for their respective deactivating enzymes. Unlike AEA and 2-AG, the novel endocannabinoid probes reported herein do not generate arachidonic acid which exhibits non-CB-mediated biological actions and in vitro and in vivo toxicity.
Acknowledgements: Research supported by NIH/NIDA grants DA009158 and DA007215.
Acknowledgements: Research supported by NIH/NIDA grants DA009158 and DA007215.
Vsevolod “Seva” Katritch
USC Michelson Center for Convergent Bioscience, Department of Quantitative and Computational Biology, Department of Chemistry, University of Southern California
USC Michelson Center for Convergent Bioscience, Department of Quantitative and Computational Biology, Department of Chemistry, University of Southern California
In the past decade, GPCR drug discovery has been transformed by the rapidly growing availability of 3D structures and a better understanding of atomistic mechanisms of signaling. This has been especially notable for the key receptors in pain and addiction disorders, including opioid and cannabinoid families, where all receptors have been structurally characterized in both inactive and active states. This talk will describe how these structures and new structure-based computational approaches enable discovery of new GPCR ligands and lead series with improved functional and safety profiles for pain and addiction disorders.
The first technology, V-SYNTHES, enables rapid identification of novel chemotypes for GPCR hits and leads in Giga-scale REadily AvaiLable (REAL) libraries of drug-like compounds (Sadybekov et al Nature, 601, 452-459) This iterative synthon-based virtual screening technology has been validated by the prospective discovery of novel antagonists for Cannabinoid (CB) from the 11 Billion compounds of REAL Space. Chemical synthesis and experimental testing of 60 compounds predicted by V-SYNTHES identified 14 novel sub-micromolar antagonists. Optimization of the best lead series by a simple SAR-by-catalog screen in the same REAL Space identified CB2 selective antagonists with nanomolar potency and 200-fold CB2/CB1 selectivity. The approach also shows promising results for other GPCRs and other classes of therapeutic targets. The next generation, fully automated V-SYNTHES2.0 is being also developed to include rapidly growing REAL Space (currently 29 Billion compounds), potentially expanding to Tera-Scale (1012-1015 compounds) screening in the next few years.
A synergistic rational design approach employs structure-guided bitopic derivatization of existing high-affinity ligand scaffolds to design new functional properties. In one example, the approach was used to discover bitopic ligands targeting both orthosteric pockets and the allosteric sodium ion binding pocket in Class A GPCRs. Because this highly conserved site deep in the 7TM helical bundle is a key part of the Class A GPCR activation mechanism, the bitopic ligands acquire new functional properties. In the application of this approach to opioid receptors, we show that the bitopic extension can differentially modulate signaling towards specific functional pathways, involving distinct G-protein subtypes. Moreover, the functionally selective fentanyl-based design demonstrated effective analgesia without respiratory depression and other opioid side effects (Faouzi et al. (2022) In revision), while the approach is being tested on other receptors. Further examples of rational scaffold functionalization, including photoswitchable groups for optical control of receptors and covalent warheads for superpotent and long-lasting ligands will be be discussed.
The first technology, V-SYNTHES, enables rapid identification of novel chemotypes for GPCR hits and leads in Giga-scale REadily AvaiLable (REAL) libraries of drug-like compounds (Sadybekov et al Nature, 601, 452-459) This iterative synthon-based virtual screening technology has been validated by the prospective discovery of novel antagonists for Cannabinoid (CB) from the 11 Billion compounds of REAL Space. Chemical synthesis and experimental testing of 60 compounds predicted by V-SYNTHES identified 14 novel sub-micromolar antagonists. Optimization of the best lead series by a simple SAR-by-catalog screen in the same REAL Space identified CB2 selective antagonists with nanomolar potency and 200-fold CB2/CB1 selectivity. The approach also shows promising results for other GPCRs and other classes of therapeutic targets. The next generation, fully automated V-SYNTHES2.0 is being also developed to include rapidly growing REAL Space (currently 29 Billion compounds), potentially expanding to Tera-Scale (1012-1015 compounds) screening in the next few years.
A synergistic rational design approach employs structure-guided bitopic derivatization of existing high-affinity ligand scaffolds to design new functional properties. In one example, the approach was used to discover bitopic ligands targeting both orthosteric pockets and the allosteric sodium ion binding pocket in Class A GPCRs. Because this highly conserved site deep in the 7TM helical bundle is a key part of the Class A GPCR activation mechanism, the bitopic ligands acquire new functional properties. In the application of this approach to opioid receptors, we show that the bitopic extension can differentially modulate signaling towards specific functional pathways, involving distinct G-protein subtypes. Moreover, the functionally selective fentanyl-based design demonstrated effective analgesia without respiratory depression and other opioid side effects (Faouzi et al. (2022) In revision), while the approach is being tested on other receptors. Further examples of rational scaffold functionalization, including photoswitchable groups for optical control of receptors and covalent warheads for superpotent and long-lasting ligands will be be discussed.
Daniel Rosenbaum
Department of Molecular Physics, UT Southwestern Medical Center
Department of Molecular Physics, UT Southwestern Medical Center
We have used different structural biology tools to characterize the atomic structure of the cannabinoid receptor CB1. Structures of this GPCR in the inactive conformation have revealed the molecular basis for binding and inhibition by inverse agonists and negative allosteric modulators. Comparison to structures in the active conformation show how CB1 responds to agonists to activate G protein signaling. This atomic view of the mechanism of CB1 is a useful framework for drug design and characterization.
Lawrence M. Carey1,2, Zhili Xu2, Gabriela Rajic2, Alexandros Makriyannis3, Julian Romero4, Cecilia Hillard5, Ken Mackie1,2,6 and Andrea G. Hohmann1,2,6*
1Program in Neuroscience, Indiana University, Bloomington, IN, 2Department of Psychological and Brain Sciences, Indiana University, Bloomington, IN, 3Center for Drug Discovery, Northeastern University, Boston, MA, 4Faculty of Experimental Sciences, Universidad Francisco de Vitoria, Madrid, Spain, 5Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, 6Gill Center for Biomolecular Science, Indiana University, Bloomington, IN
1Program in Neuroscience, Indiana University, Bloomington, IN, 2Department of Psychological and Brain Sciences, Indiana University, Bloomington, IN, 3Center for Drug Discovery, Northeastern University, Boston, MA, 4Faculty of Experimental Sciences, Universidad Francisco de Vitoria, Madrid, Spain, 5Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, 6Gill Center for Biomolecular Science, Indiana University, Bloomington, IN
Painful peripheral neuropathy is the most common neurological complication associated with human immune deficiency virus (HIV) infection. Currently available treatments fail to provide adequate symptom relief, indicating the need for novel treatment strategies. To address this gap in knowledge, we characterized the impact of cannabinoid CB2 agonists, which lack psychoactivity associated with central CB1 activation, on antiretroviral-induced neuropathic nociception and identified cell types expressing CB2 that mediate the antinociceptive efficacy of CB2 agonists. Two structurally distinct CB2 agonists (AM1710 and LY2828360) alleviated antiretroviral-induced neuropathic pain, benefits which were absent in CB2 knockout mice. Conditional deletion of CB2 from peripheral sensory neurons eliminated the antinociceptive efficacy of CB2 agonists. We also asked whether LY2828360 treatment could reverse established morphine tolerance in the ddC-induced neuropathy model and whether CB2 expression on peripheral sensory neurons is necessary for sparing of morphine tolerance by LY2828360. The present studies suggest that CB2 activation may alleviate HIV-associated antiretroviral neuropathy and identify a previously unreported mechanism through which CB2 activation produces antinociceptive efficacy. Our results also provide the first evidence that a CB2 agonist can reverse established morphine tolerance and demonstrate that CB2 localized to peripheral sensory neurons mediates the opioid tolerance sparing efficacy of CB2 agonists.
This work was supported by National Institute on Drug Abuse Grants DA047858, DA041229 (A.G.H. and K.M.), and DA042584 (A.G.H.), National Cancer Institute Grant CA200417 (A.G.H.), an Indiana Addiction Grand Challenge Grant (A.G.H.), the Research and Education Component of the Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin (C.J.H) and the Ministerio de Economía y Competitividad (SAF 2016-75959-R and SAF PID2019-108992RB-I00 to JR). L.M.C. was supported by National Institute on Drug Abuse T32 training grant DA024628 and the Harlan Scholars Research Program.
This work was supported by National Institute on Drug Abuse Grants DA047858, DA041229 (A.G.H. and K.M.), and DA042584 (A.G.H.), National Cancer Institute Grant CA200417 (A.G.H.), an Indiana Addiction Grand Challenge Grant (A.G.H.), the Research and Education Component of the Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin (C.J.H) and the Ministerio de Economía y Competitividad (SAF 2016-75959-R and SAF PID2019-108992RB-I00 to JR). L.M.C. was supported by National Institute on Drug Abuse T32 training grant DA024628 and the Harlan Scholars Research Program.
Hideaki Yano
Bouvé College of Health Sciences, School of Pharmacy, Department of Pharmaceutical Sciences, Center for Drug Discovery, Northeastern University
Misuse of synthetic cannabinoid receptor agonists (SCRAs) has risen worldwide and is often associated with serious adverse effects including hallucinations, cardiovascular abnormalities, coma, or death. Many SCRAs are aminoalkylindole molecules that are distinct from the naturally occurring psychoactive phytocannabinoid, Δ9-tetrahydrocannabinol (Δ9-THC), found in cannabis. It is established that Δ9-THC is a partial agonist at cannabinoid CB1 receptors, whereas SCRAs demonstrate much higher efficacy and potency at this site, and this likely relates to a much higher incidence of medically adverse effects. Moreover, some SCRAs exhibit a maximal efficacy at CB1 receptors exceeding typical full agonists, and this has been termed “superagonism.” Additionally, superagonism is usually associated with increased potency at CB1 receptors. At present, it is unknown whether these unique pharmacological characteristics at CB1 receptors directly relate to the adverse effects of some SCRAs, or whether their actions at non-CB1 sites may also be involved. In the current study, we examined molecular mechanisms of superagonism of SCRAs at CB1 receptors.
We first tested a series of SCRAs in our bioluminescence resonance energy transfer (BRET) assay in a CB1-Gi protein engagement configuration. Unlike other receptor function assays, this approach avoids caveats associated with signal amplification that can lead to false-positives for superagonism. Based on the structure-activity relationship for superagonism established by this BRET assay, we then performed molecular dynamics simulations of the CB1 receptor complexed with selected SCRAs to investigate the molecular determinants of SCRA binding efficacy, and to characterize superagonism conformational states of the CB1 receptor-SCRA complex. Finally, to evaluate superagonism of selected SCRAs at CB1 receptors in the mouse brain, we performed electrophysiological experiments in hippocampal brain slices to determine effects of these SCRAs on glutamate release inhibition mediated by CB1 receptors. In short, our findings delineate molecular mechanisms of superagonism at CB1 receptors and further our understanding of the mechanisms through which SCRAs may cause serious adverse health effects.
We first tested a series of SCRAs in our bioluminescence resonance energy transfer (BRET) assay in a CB1-Gi protein engagement configuration. Unlike other receptor function assays, this approach avoids caveats associated with signal amplification that can lead to false-positives for superagonism. Based on the structure-activity relationship for superagonism established by this BRET assay, we then performed molecular dynamics simulations of the CB1 receptor complexed with selected SCRAs to investigate the molecular determinants of SCRA binding efficacy, and to characterize superagonism conformational states of the CB1 receptor-SCRA complex. Finally, to evaluate superagonism of selected SCRAs at CB1 receptors in the mouse brain, we performed electrophysiological experiments in hippocampal brain slices to determine effects of these SCRAs on glutamate release inhibition mediated by CB1 receptors. In short, our findings delineate molecular mechanisms of superagonism at CB1 receptors and further our understanding of the mechanisms through which SCRAs may cause serious adverse health effects.
Madelyn H. Ray, BaDoi Phan, Marianne L. Seney, Jill R. Glausier, Allison Tipton, Shelley J. Russek, Andreas Pfenning, David A. Lewis, and Ryan W. Logan
Pharmacology and Experimental Therapeutics, Boston University School of Medicine
Pharmacology and Experimental Therapeutics, Boston University School of Medicine
In the United States, rates of opioid use disorder (OUD) and deaths from overdose are unprecedented. Despite the enormous public health impact of OUD, we have a limited understanding of the changes in the brain in patients with OUD. Human neuroimaging studies investigating OUD have implicated functional changes in the dorsal striatum, an area integral in reward processing, habitual drug-seeking, craving, and relapse. Few studies have directly examined the cellular and molecular changes in the human brain associated with OUD. Here, we collected caudate nucleus and putamen, subregions of the dorsal striatum, from postmortem brains of unaffected comparison subjects (n=12) and subjects with OUD (n=12). Both caudate and putamen were processed for single nuclei RNA-seq. Nuclei were sorted using flow cytometry, then barcoded and processed using the 10X Genomics pipeline. We sequenced 7,000 nuclei per sample at an average depth of 50,000 reads per nuclei. We identified transcriptional alterations associated with OUD in specific cell types of the striatum, including significant shifts in the expression of dopamine receptor subtypes in GABAergic medium spiny neurons. Additional exploratory approaches are currently underway to reveal cell-specific alterations in gene expression and compare different cell types across striatal subregions. Our results are the first to demonstrate significant alterations in gene expression across different cell types of the striatum in the human brain associated with OUD using single nuclei RNA-seq. Uncovering molecular mechanisms of opioid use will propel the identification of new therapeutic targets and the development of successful treatment strategies for OUD.
Bryan Roth
Department of Pharmacology, University of North Carolina
Department of Pharmacology, University of North Carolina
I will present new and as yet unpublished data regarding the structure, function and pharmacology of MRGPRX-family receptors.
Takashi Tsukamoto
Johns Hopkins Drug Discovery Program, Johns Hopkins University
Mas-related G-protein coupled receptor X1 (MRGPRX1) is a human sensory neuron-specific receptor and has gained increasing interest as a therapeutic target for the treatment of pain. Positive allosteric modulators (PAMs) of MRGPRX1 represent a promising new class of agents to treat pain by preferentially activating the receptors at the central terminals of primary sensory neurons, minimizing itch side effects caused by the activation of these receptors on the peripheral nerves. Our collaborative efforts supported by NIH’s HEAL Initiative towards developing potent, orally available, thieno[2,3-d]pyrimidine-based MRGPRX1 PAMs will be presented.
Randal A. Serafinia*, Justin J. Frereb,c*, Jeffrey Zimeringa,d, Kerri D. Prycea, Ilinca M. Giosan, Ilona Golynkerc, Maryline Panisc, Anne Ruiza, Benjamin tenOeverc°, Venetia Zacharioua,e°
a Nash Department of Neuroscience and Friedman Brain Institute, Icahn School of Medicine at Mount Sinai; b Department of Microbiology, Icahn School of Medicine at Mount Sinai; c Department of Microbiology, New York University Langone; d Department of Neurosurgery, Icahn School of Medicine at Mount Sinai; e Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai
a Nash Department of Neuroscience and Friedman Brain Institute, Icahn School of Medicine at Mount Sinai; b Department of Microbiology, Icahn School of Medicine at Mount Sinai; c Department of Microbiology, New York University Langone; d Department of Neurosurgery, Icahn School of Medicine at Mount Sinai; e Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai
Despite being largely confined to the airways, SARS-CoV-2 infection has been associated with sensory abnormalities that manifest in both acute and long-lasting phenotypes. To gain insight on the molecular basis of these sensory abnormalities, we used the golden hamster infection model to characterize the effects of SARS-CoV-2 versus Influenza A virus (IAV) infection on the sensory nervous system. Efforts to detect the presence of virus in the cervical/thoracic spinal cord and dorsal root ganglia (DRGs) demonstrated detectable levels of SARS-CoV-2 by quantitative PCR and RNA scope uniquely within the first 24 hours of infection. SARS-CoV-2-infected hamsters demonstrated mechanical hypersensitivity during acute infection; intriguingly, this hypersensitivity was milder, but prolonged when compared to IAV-infected hamsters. RNA sequencing (RNA-seq) of thoracic DRGs from acute infection revealed predominantly neuron-biased signaling perturbations in SARS-CoV-2-infected animals as opposed to type I interferon signaling in tissue derived from IAV-infected animals. RNA-seq of 31dpi thoracic DRGs from SARS-CoV-2-infected animals highlighted a uniquely neuropathic transcriptomic landscape, which was consistent with substantial SARS-CoV-2-specific mechanical hypersensitivity at 28dpi. Ontology analysis of 1, 4, and 30dpi RNA-seq revealed novel targets for pain management, such as ILF3. Meta-analysis of all SARS-CoV-2 RNA-seq timepoints against preclinical pain model datasets highlighted both conserved and unique pro-nociceptive gene expression changes following infection. Overall, this work elucidates novel transcriptomic signatures triggered by SARS-CoV-2 that may underlie both short- and long-term sensory abnormalities while also highlighting several therapeutic targets for alleviation of infection-induced hypersensitivity.
S. Stevens Negus
Department of Pharmacology and Toxicology, Virginia Commonwealth University
Department of Pharmacology and Toxicology, Virginia Commonwealth University
The opioid epidemic has stimulated efforts to discover new treatments for opioid use disorder (OUD); however, since the approval of buprenorphine in 2002, no new OUD medications have attained approval by the U. S. Food and Drug Administration. One obstacle to OUD medication development has been an excessive reliance on preclinical testing procedures that yield a high rate of false positive effects. Preclinical-to-clinical predictive validity can be improved with the use of drug-choice procedures, in which subjects choose between a drug reinforcer (e.g. an opioid like fentanyl or heroin) and a concurrently available non-drug reinforcer (e.g. food in laboratory animals or money in humans). In these procedures, promising medications not only produce a decrease in drug choice, but also promote a reallocation of behavior to choice of the alternative non-drug reinforcer. Less promising candidates fail to produce this outcome, and instead often have little effect on drug choice while depressing overall rates of responding and reinforcement. This talk will describe drug choice procedures developed for use in nonhuman primates and rats, describe effects of both effective and ineffective treatments, and highlight the importance of opioid dependence/withdrawal state on medication effects.
Lipin Ji1, Anna W. Sromek2, Thanh Ho2, Kiran Vemuri2, Shakiru Alapafuja1, Alexander Zvonok1, Alexandros Makriyannis2*
1MAKScientific 2Center for Drug Discovery and Department of Pharmaceutical Sciences, Northeastern University
The endocannabinoid system is involved in mediating the rewarding effects of drugs of abuse. Blockade the CB1 receptor represents an effective approach toward treating substance use disorders. First generation CB1 antagonists (i.e., rimonabant) possessed inverse agonist activity, and led to unwanted psychiatric side effects. Second generation CB1 antagonists were developed to functionally act as neutral antagonists, which notably lack inverse agonist activity and their attendant unwanted side effects. This talk will focus on ongoing efforts to develop AM6527, a centrally acting CB1 neutral antagonist, as a medication for treating alcohol use disorder. The potential to develop CB1 neutral antagonists as medications for other substance use disorders will also be discussed.
Support from the NIH, NIAAA grant U01 AA028963 is gratefully acknowledged.
Kimberly N. Karin1*, Erica N. Golden1, Raphael Mechoulam2, Linda Parker3, M. Imad Damaj1,
Aron H. Lichtman1, and Joel E. Schlosburg1
1Dept of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, VA, USA 2Institute of Drug Research, Medical Faculty, Hebrew University of Jerusalem, Jerusalem, Israel 3Dept of Psychology and Collaborative Neuroscience Program, University of Guelph, Guelph,2ON, N1G 2W1, Canada
1Dept of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, VA, USA 2Institute of Drug Research, Medical Faculty, Hebrew University of Jerusalem, Jerusalem, Israel 3Dept of Psychology and Collaborative Neuroscience Program, University of Guelph, Guelph,2ON, N1G 2W1, Canada
Introduction: With over 16 million individuals in the United States suffering from tobacco-related diseases, tobacco dependence continues to burden the healthcare system despite a majority of smokers reporting the desire to quit. Current nicotine cessation pharmacotherapies have limited efficacy in supporting long-term cessation of tobacco and nicotine. As nicotine dependence continues to be a major public health issue, new nicotine cessation pharmacotherapies are needed. Our group previously reported that the novel, endogenous compound n-oleoyl glycine (OlGly) reduced nicotine reward, as assessed in a conditioned place paradigm (CPP), through a peroxisome proliferator activated receptor-alpha (PPAR-α) dependent mechanism. Other studies suggest that the structural analogue of OlGly, N-oleoyl alanine (OlAla), has an increased duration of action, which would be of benefit from a translation perspective. In the present study, we assessed OlGly and OlAla in the rat models of nicotine seeking: self-administration and cue/prime-induced reinstatement.
Methods: Male Wistar rats implanted with jugular catheters were trained to self-administer 0.03 mg/kg nicotine infusions on a fixed-ratio (FR) 2 schedule during daily 2 hr sessions. OlGly, OlAla (5, 10, 20 mg/kg), or vehicle (1:1:8 ethanol:emulphor:saline) were administered by intraperitoneal (ip) injection 15 mins prior to the sessions. In order to elucidate the potential underlying mechanisms of these drugs, subjects were pretreated with the PPAR-α antagonist GW6471 (2 mg/kg), the PPAR-γ antagonist GW9662 (2 mg/kg), or the CB1 receptor antagonist rimonabant (0.3 mg/kg). For reinstatement studies, lever responding behavior was extinguished by removing paired cues and nicotine infusions. A priming dose of nicotine (0.3 mg/kg; subcutaneous) in combination with cue presentation produced robust reinstatement. Subjects were given an ip injection of OlGly or OlAla (5, 10, 15 mg/kg) 10 mins prior to administration of the nicotine priming dose.
Results: Both OlGly and OlAla significantly reduced nicotine lever responding. Compared to responding when a pretreatment of vehicle was administered, 10 and 20 mg/kg OlGly reduced responding to 76% and 39%, respectively. OlAla (10 and 20 mg/kg) reduced nicotine responding to 78% and 50%, respectively, compared to vehicle pretreatment responding. Of the three antagonists tested, only GW6471 blocked OlGly-induced reductions in nicotine lever responding. In contrast, none of the antagonists prevented the effects of OlAla. In addition to reducing nicotine self-administration, both OlGly and OlAla significantly reduced cue/prime-induced reinstatement. OlGly (10 and 15 mg/kg) respectively reduced reinstatement to 49% and 33% of the vehicle reinstatement response rates. Likewise, OlAla (10 and 15 mg/kg) respectively reduced reinstatement responses to 59% and 44% of vehicle rates. Conclusions: OlGly and OlAla reduced nicotine seeking in both rat self-administration and cue/prime-induced reinstatement paradigms. While the observation OlGly reduced nicotine self-administration through a that PPAR-α mechanisms of action aligns with our previous report using the mouse model of nicotine-induced CPP, OlAla reduces nicotine reward through a distinct mechanism. Collectively, these findings indicate that the structurally similar lipid signaling molecules OlGly and OlAla hold promise as potential nicotine cessation pharmacotherapies.
Acknowledgements: Funded by National Institute of Health (P30DA033934 and T32DA007027) as well as by the PlantEXT LTD drug development program under pending patents.
Methods: Male Wistar rats implanted with jugular catheters were trained to self-administer 0.03 mg/kg nicotine infusions on a fixed-ratio (FR) 2 schedule during daily 2 hr sessions. OlGly, OlAla (5, 10, 20 mg/kg), or vehicle (1:1:8 ethanol:emulphor:saline) were administered by intraperitoneal (ip) injection 15 mins prior to the sessions. In order to elucidate the potential underlying mechanisms of these drugs, subjects were pretreated with the PPAR-α antagonist GW6471 (2 mg/kg), the PPAR-γ antagonist GW9662 (2 mg/kg), or the CB1 receptor antagonist rimonabant (0.3 mg/kg). For reinstatement studies, lever responding behavior was extinguished by removing paired cues and nicotine infusions. A priming dose of nicotine (0.3 mg/kg; subcutaneous) in combination with cue presentation produced robust reinstatement. Subjects were given an ip injection of OlGly or OlAla (5, 10, 15 mg/kg) 10 mins prior to administration of the nicotine priming dose.
Results: Both OlGly and OlAla significantly reduced nicotine lever responding. Compared to responding when a pretreatment of vehicle was administered, 10 and 20 mg/kg OlGly reduced responding to 76% and 39%, respectively. OlAla (10 and 20 mg/kg) reduced nicotine responding to 78% and 50%, respectively, compared to vehicle pretreatment responding. Of the three antagonists tested, only GW6471 blocked OlGly-induced reductions in nicotine lever responding. In contrast, none of the antagonists prevented the effects of OlAla. In addition to reducing nicotine self-administration, both OlGly and OlAla significantly reduced cue/prime-induced reinstatement. OlGly (10 and 15 mg/kg) respectively reduced reinstatement to 49% and 33% of the vehicle reinstatement response rates. Likewise, OlAla (10 and 15 mg/kg) respectively reduced reinstatement responses to 59% and 44% of vehicle rates. Conclusions: OlGly and OlAla reduced nicotine seeking in both rat self-administration and cue/prime-induced reinstatement paradigms. While the observation OlGly reduced nicotine self-administration through a that PPAR-α mechanisms of action aligns with our previous report using the mouse model of nicotine-induced CPP, OlAla reduces nicotine reward through a distinct mechanism. Collectively, these findings indicate that the structurally similar lipid signaling molecules OlGly and OlAla hold promise as potential nicotine cessation pharmacotherapies.
Acknowledgements: Funded by National Institute of Health (P30DA033934 and T32DA007027) as well as by the PlantEXT LTD drug development program under pending patents.
Emily L. Burke1, Paul T. Bremer2, Rajeev I. Desai1
1Integrative Neurochemistry Laboratory, McLean Hospital, Harvard Medical School, Belmont, MA, 2Cessation Therapeutics, San Diego, CA
1Integrative Neurochemistry Laboratory, McLean Hospital, Harvard Medical School, Belmont, MA, 2Cessation Therapeutics, San Diego, CA
Fentanyl is a synthetic opioid that is routinely abused for its pleasurable psychoactive effects, which can lead to substance use disorder (SUD) and subsequent overdose. Although treatments for SUD exist, like opioid replacement therapy with μ-opioid receptor agonists like methadone or buprenorphine, they can be ineffective and have adverse side-effects. Thus, antibody-based therapeutics have emerged as a new approach to treating SUD due to their specificity, low toxicity, and immediate effects post-infusion. The present experiments were conducted to examine the ability of a novel fentanyl-targeting monoclonal antibody, CSX-1004, to block fentanyl’s respiratory depressant effects. Eight squirrel monkeys were treated with either 10 or 40 mg/kg CSX-1004 via IV infusion and were cumulatively given fentanyl on days 0, 2, 7, 14, 21, and 28 post-infusion. Four subjects also received a second infusion of 40 mg/kg CSX-1004 after a 2-month washout period to determine the magnitude of fentanyl antagonism. Respiratory depression was examined using a ventilation chamber in which subjects were exposed to air and air with 5% CO2 in 10-minute alternating increments. The volume of breaths per minute was measured and used to generate dose-response functions. CSX-1004 successfully blocked fentanyl’s respiratory depressant effects in both a dose- and time-dependent manner. Fentanyl challenge on Day 0 post-CSX-1004 infusion resulted in no fentanyl-associated reductions in minute volume, and these effects continued through Day 28, although efficacy gradually diminished starting at Day 14. Subjects that received the 10 mg/kg dose returned to baseline levels faster than those that received the 40 mg/kg dose, with efficacy beginning to diminish at day 7. The 40 mg/kg CSX-1004 dose also produced a significant >30-fold rightward shift in the dose-response function of fentanyl. These initial studies suggest that monoclonal antibodies could be a promising treatment in preventing fentanyl overdose.
Yiorgos Skiniotis
Department of Molecular and Cellular Physiology, Department of Structural Biology, Stanford University
Department of Molecular and Cellular Physiology, Department of Structural Biology, Stanford University
Drugs targeting the μ-opioid receptor (μOR) are the most effective analgesics but are associated with the severe side effects behind the current opioid epidemic. Here I will present our work on the structural and mechanistic basis of action of µOR agonists with different safety profiles. I will discuss how cryo-EM structures, molecular dynamics simulations and cell-based assays suggest that drugs engaging different parts of the μOR orthosteric pocket may lead to distinct signaling outcomes.
Gregory Dudley
C. Eugene Bennett Department of Chemistry, West Virginia University
C. Eugene Bennett Department of Chemistry, West Virginia University
Highly substituted benzenes — like the penta- or hexasubstituted benzene cores of the illudalane sesquiterpenes — are underrepresented in medicinal chemistry and present long-standing challenges to chemical synthesis. Traditional synthetic approaches feature serial aromatic substitution, which becomes increasingly cumbersome with increasing substitution, rendering many substituted benzenes inaccessible for comprehensive pharmacological assessment. Despite these challenges, interest in the illudalanes and related compounds has surged in recent years, highlighted by the discovery that illudalic acid (from the toxic jack o’lantern mushroom Omphalotus illudens) is a potent and selective inhibitor of the LAR family of protein tyrosine phosphatases (PTPs). We will describe benzannulation methodologies and convergent syntheses of illudalic acid and illudalane-type structures aimed at advancing the pharmacology of drug abuse, including LAR/PTPRD inhibitors of interest for addiction research and a novel CB2-selective indole cannabinoid antagonist.
Christos Iliopoulos-Tsoutsouvas
Center for Drug Discovery and Department of Pharmaceutical Sciences, Northeastern University, Boston
The two cannabinoid receptors, CB1 and CB2, are involved in many pathological processes and present attractive therapeutic targets. Recently, the ligand-bound structures of the cannabinoid receptors have been determined in high resolution using X-ray and cryo-EM approaches. The available ligand-receptor complexes, to date, have been elucidated in the presence of several agonists, antagonists, and inverse agonists, that belong to various structurally distinct classes. These structures uncovered features of the activation/deactivation mechanisms, ligand recognition elements, and their implications for drug discovery. This talk will focus on the orthosteric binding pockets of CB1 and CB2, what has been learned so far and what is more to learn regarding subtype selectivity and the functional profiles of several cannabinoid ligands. Also, comparing the CB1 and CB2 structures indicates that it is possible to obtain dual-acting ligands with opposing functions between the two receptors. The knowledge obtained sets the basis for the efficient drug discovery of cannabinoid ligands.
Christoph Gorgulla
Harvard Medical School and Department of Physics, Harvard University
Harvard Medical School and Department of Physics, Harvard University
On average, an approved drug currently costs US$2–3 billion and takes more than 10 years to develop. In part, this is due to expensive and time-consuming wet-laboratory experiments, poor initial hit compounds and the high attrition rates in the (pre-)clinical phases. These challenges also affect small-molecule drug development projects aiming to reverse addiction and prevent drug abuse. Structure-based virtual screenings have the potential to mitigate the above mentioned problems. Within structure-based virtual screenings, the quality of the hits improves with the number of compounds screened. In this talk I will describe VirtualFlow, a highly automated and versatile drug discovery platform with perfect scaling behaviour that is able to prepare and efficiently screen ultra-large libraries of compounds. VirtualFlow was the first platform able to routinely screen billions of commercially available molecules.
Ryan McGlynn1, Meng Cui1, Brittany Brems1, Stephen Kohut3, Alex Yuan1, Raymond G. Booth1,2
1 Department of Pharmaceutical Sciences, Northeastern University, Boston MA, 2 Department of Chemistry, Northeastern University, Boston MA, 3 Department of Physiatry, McLean Hospital, Belmont MA
Serotonin (5-Hydroxytyrtrptamine, 5-HT) systems putatively play a role in pathophysiology and pharmacotherapy of anxiety, depression, sociability, and obsessive compulsion disorder, and substance use disorder (SUD). The 5-HT1R family, which comprises of the 5-HT1A/1B/1D/1e/1FR G-protein coupled receptor (GPCR) subtypes, is the largest 5-HT receptor family. In a clinical setting, 5-HT1R agonists, such as buspirone, have shown efficacy to treat heroin withdrawal symptoms.
Our lab has developed novel analogs of the chiral 5-subsitituted 2-aminotetralin (5-SAT) chemotype that bind to members of the 5-HT1R family. However, 5-HT1R subtypes share ~80% binding pocket amino acid homology, making development of receptor specific ligands a challenge. To understand the molecular pharmacology of 5-SATs at 5-HT1Rs we are studying substituents at the C(2)-chiral amine and the C(5) positions and their effect on affinity (Ki) and functional potency (EC50) and efficacy (EMax) to impact adenylyl cyclase induced cAMP formation in HEK293t cells transiently expressing each of the human 5-HT1R subtypes. Preliminary results indicate that the chemistry of the substituent at the C(2) and C(5) positions of the 5-SAT scaffold affects the Ki, EC50, and Emax differentially at 5-HT1 subtypes. In general, it was found that 5-SATs have potent affinity at 5-HT1A/1B/1DR subtypes with Ki values < 5nM at specific subtypes depending on the C(2) and C(5) substituent but have nil affinity (Ki >5μM) at the 5-HT1FR. The 5-SATs assessed all were agonists at 5-HT1A/1B/1DRs, with EC50 values under 5nM and Emax values spanning 25-100% of the maximal response of full agonist 5-CT at specific subtypes depending on the C(2) and C(5) substituent. Molecular modeling and molecular dynamics (MD) studies inform how 5-SATs bind and function at 5-HT1A/1BR subtypes and provide framework for binding pose validation by site-directed mutagenesis studies. Lead 5-SATs that selectively activate 5-HT1AR over the 5-HT1BR (~30-Fold EC50 difference) were docked at models of the 5-HT1AR cyro-EM structure (PBD = 7E2Y) and the 5-HT1BR crystal structure (PDB =4IAR). MD studies have illuminated key halogen-hydroxyl interactions between compound moieties and transmembrane domains 5 and 6 that drive functional potency differences at 5-HT1A vs. 5-HT1B receptors. Observations from MD studies will inform structure activity relationships for 5-SATs at the 5-HT1R subtypes to guide medicinal chemical optimization for drug development. Furthermore, the Kohut group at McLean Hospital showed that administration of novel 5-SAT analogs can reduce withdrawal symptoms (i.e., lying on side, holding abdomen, grimacing, pacing, crawling, retching, meiosis, and dysphoric facial expression) in a cohort of rhesus monkeys addicted to morphine.
Our lab has developed novel analogs of the chiral 5-subsitituted 2-aminotetralin (5-SAT) chemotype that bind to members of the 5-HT1R family. However, 5-HT1R subtypes share ~80% binding pocket amino acid homology, making development of receptor specific ligands a challenge. To understand the molecular pharmacology of 5-SATs at 5-HT1Rs we are studying substituents at the C(2)-chiral amine and the C(5) positions and their effect on affinity (Ki) and functional potency (EC50) and efficacy (EMax) to impact adenylyl cyclase induced cAMP formation in HEK293t cells transiently expressing each of the human 5-HT1R subtypes. Preliminary results indicate that the chemistry of the substituent at the C(2) and C(5) positions of the 5-SAT scaffold affects the Ki, EC50, and Emax differentially at 5-HT1 subtypes. In general, it was found that 5-SATs have potent affinity at 5-HT1A/1B/1DR subtypes with Ki values < 5nM at specific subtypes depending on the C(2) and C(5) substituent but have nil affinity (Ki >5μM) at the 5-HT1FR. The 5-SATs assessed all were agonists at 5-HT1A/1B/1DRs, with EC50 values under 5nM and Emax values spanning 25-100% of the maximal response of full agonist 5-CT at specific subtypes depending on the C(2) and C(5) substituent. Molecular modeling and molecular dynamics (MD) studies inform how 5-SATs bind and function at 5-HT1A/1BR subtypes and provide framework for binding pose validation by site-directed mutagenesis studies. Lead 5-SATs that selectively activate 5-HT1AR over the 5-HT1BR (~30-Fold EC50 difference) were docked at models of the 5-HT1AR cyro-EM structure (PBD = 7E2Y) and the 5-HT1BR crystal structure (PDB =4IAR). MD studies have illuminated key halogen-hydroxyl interactions between compound moieties and transmembrane domains 5 and 6 that drive functional potency differences at 5-HT1A vs. 5-HT1B receptors. Observations from MD studies will inform structure activity relationships for 5-SATs at the 5-HT1R subtypes to guide medicinal chemical optimization for drug development. Furthermore, the Kohut group at McLean Hospital showed that administration of novel 5-SAT analogs can reduce withdrawal symptoms (i.e., lying on side, holding abdomen, grimacing, pacing, crawling, retching, meiosis, and dysphoric facial expression) in a cohort of rhesus monkeys addicted to morphine.
David E. Nichols
Department Of Pharmacology, Eshelman School of Pharmacy, University of North Carolina
This talk will begin with a brief review of the natural sources of psychedelics that have been used for millennia. That will be followed by a discussion of the 5-HT2A receptors that are the targets for the classic psychedelics; where they are located and very brief comments on the nature and function of G protein-coupled receptors. There will be brief discussion of some preclinical studies from the Nichols lab, focused on elucidation of the conformation of the diethylamide function of LSD. In particular, Leucine 229, in extracellular loop 2 of the receptor was shown to be a critical residue in the receptor kinetics of LSD binding to the receptor. A few of the groundbreaking projects supported by the Heffter Research Institute, founded by the speaker in 1993, will be mentioned. Finally, unpublished structures of several molecules bound to the human 5-HT2A receptor will be shown and very briefly noted.
Albert Garcia-Romeu
Department of Psychiatry and Behavioral Sciences
Johns Hopkins University School of Medicine
Early research examined use of psychedelic drugs such as LSD in the treatment of various health conditions including for palliative care and addictions. Research in this area was largely halted in the 1970s after criminalization and due to stigma associated with widespread recreational use. The 21st century has seen a renewed interest in this area with psychedelics such as psilocybin and ayahuasca being studied again for potential therapeutic effects in anxiety, mood, and substance use disorders. This presentation will provide a historical overview of clinical research with psychedelics and discuss recent developments in the field. Recent preliminary studies investigating mental health outcomes of psychedelic-assisted treatments will be presented and potential biological and psychological mechanisms will be described in brief.
Mahmoud Nasr
Renal Division, Brigham and Women’s Hospital, Harvard Medical School
Renal Division, Brigham and Women’s Hospital, Harvard Medical School
Viral infections represent a major health threat to humankind. We will present a new detection technology which has the means to potentially provide an instant detection of many viruses. We engineered biosensors that assemble into active enzyme in the presence of viral surface proteins from HIV, Influenza and SARS-CoV-2. Given the current need for diagnostic tools for SARS-CoV-2, we focused our efforts in optimizing our approach to achieve the high sensitivity required to detect SARS-CoV-2 clinically. We validated our detection method using the saliva samples from COVID-19 patients. Our approach is well-suited to substantially reduce the complexity and improve the scalability for the point-of-care diagnostics of infectious diseases.
Support from the NIH RADX Award: U54HL119145 is gratefully acknowledged.
Support from the NIH RADX Award: U54HL119145 is gratefully acknowledged.
Balazs R. Varga1, Sarah M. Bernhard1, Amal El Daibani1, Saheem Zaidi2, Kevin Appourchaux1, Samantha Harker1, Alexa Kouvelis1, Shainnel O. Eans3, Vsevolod Katritch2, Jay P. McLaughlin3, Susruta Majumdar1 and Tao Che1
1Center for Clinical Pharmacology, University of Health Sciences & Pharmacy and Washington University School of Medicine, St. Louis, MO, USA
2 Department of Quantitative & Computational Biology and Department of Chemistry, University of Southern California, Los Angeles, CA, USA.
3Department of Pharmacodynamics, University of Florida, Gainesville, FL, USA.
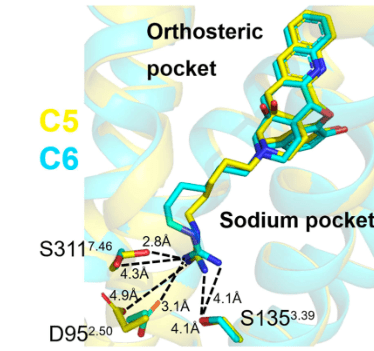
Chronic pain effects 11.2% of Americans. Opioids are used for the treatment of pain but are associated with overdoses and addiction liability, which has led to the opioid epidemic. The need for a safer opioid-based analgesic led us to target the delta opioid receptor (δOR) as an alternate to the mu opioid receptor (μOR). Using a structure-based approach we transformed a well-known δOR antagonist NTI into bitopic agonists through engagement of the sodium binding pocket, a surprising modulation of activity in view of the fact that for many Family A G protein-coupled receptors (GPCRs), the natural ligand sodium (Na+) acts as a negative allosteric modulator. Functional studies suggest that bitopic ligand interactions with the Na+ pocket control their efficacy and functional selectivity profiles for arrestins, and differential signaling can be achieved and is highly dependent on the length of the linkers used to engage the sodium binding pocket. We obtained single particle cryo-EM structures of δOR in complex with the two most promising bitopic agonists, C5-quino (2.8 Å) and C6-quino (2.8 Å), validating their design and highlighting key interactions between the guanidine group of bitopics and the Na+-binding pocket. In vivo, C6-quino displayed antinociception in a chronic pain model and was devoid of δOR related adverse effects like convulsions (seen with first generation δOR agonists) and hyperlocomotion, further strengthening the idea that targeting the Na+ site can provide an avenue for the design of safer analgesics. To the best of our knowledge this is only the second attempt to target the allosteric Na+ site (present in ~600 out of 719 Class A GPCR’s) at any GPCR which are targets for 40% of all FDA approved medications.
Wilkerson JL1,5, Zuarth Gonzalez JD1,5, Mazpule-Carrigan CC1, Patel RC1, Patel A1, Obeng S1,2, Leon F2, Mottinelli M2, Mukhopadhyay S2, Hiranita T1,5, McCurdy CR2,3,4, and McMahon LR1,5
Departments of Pharmacodynamics1, Medicinal Chemistry2, and Pharmaceutics3, and Translational Drug Development Core, Clinical and Translational Sciences Institute4, College of Pharmacy, University of Florida; Department of Pharmaceutical Sciences, Texas Tech University Health Sciences Center, Jerry H. Hodge School of Pharmacy, Texas5
Introduction: Kratom (Mitragyna speciosa), a natural product from Southeast Asia, has gained popularity in recent years and is widely available in the United States. Kratom users claim great success self-treating opioid dependence, yet scientific evidence is lagging. Amidst the current opioid epidemic finding pharmacotherapies for opioid use disorders (OUD) is a public health priority. The 40+ alkaloids within kratom serve as natural leads in the development of OUD treatments. For the current study, we examined the most abundant alkaloid in the plant: mitragynine (MG) in both self-administration and withdrawal studies. We also studied its metabolites 7-hydroxy-mitragynine (7-OH-MG) and mitragynine pseudoindoxyl (MG-P) in self-administration studies.
Methods: In one cohort (4 males and 4 females) of Sprague Dawley rats, intravenous (i.v.) self-administration for the opioid agonist, remifentanil was established during the light cycle. The experiment was conducted within subjects and each session was made up of five (30-minute) components starting under extinction condition and increasing remifentanil doses per component (0.1, 0.32, 1, 3.2 µg/kg/infusion). In these rats trained to self-administer remifentanil, the selectivity of intraperitonially (i.p.) administered test compounds (MG, 7-OH-MG, and MG-P) to antagonize responding for remifentanil vs. a non-opioid, cocaine, was assessed. The abuse potential of MG, 7-OH-MG, and MG-P was also studied within the same cohort. In a separate cohort (4 males and 4 females) of Sprague Dawley rats, rats received escalating doses of morphine (10-40 mg/kg, b.i.d.) paired with either vehicle, or MG (32, 56 mg/kg) over the course of 6 days. On day 6 all rats were given naltrexone (10 mg/kg), along with either their vehicle or MG treatment. Opioid withdrawal was assessed via counting instances of naltrexone-induced diarrhea.
Results: Remifentanil maintained self-administration above extinction levels at most doses tested (0.32, 1, and 3.2 µg/kg/infusion). Each of the kratom alkaloids, MG, 7-OH-MG, and MG-P, were 3- to 4-fold more potent to decrease remifentanil response rates (alkaloid ED50 values: 47.8, 1.99, and 3.36 µg/kg, respectively) than they were to decrease the maximum cocaine cross-administration response rates (alkaloid ED50 values: 135, 7.67, and 10.6 µg/kg, respectively). That is, the antagonistic effects of MG, 7-OH-MG, and MGP were relatively specific for the reinforcing effects of remifentanil over those of cocaine. When the test compounds were substituted for i.v. remifentanil, self-administration responding above extinction was maintained by 7-OH-MG and MGP, but not MG. MG (32 mg/kg) attenuated the development of naltrexone-induced morphine withdrawal signs. Conclusions: Overall, these results in rats suggest low MG abuse potential and support the idea that MG holds therapeutic promise as an opioid dependence medication. However, at high doses above naturally occurring MG metabolism, isolated MG active metabolites may possess abuse potential and warrant consideration for further studies.
Supported by National Institute on Drug Abuse grants DA25267 and DA048353.
Methods: In one cohort (4 males and 4 females) of Sprague Dawley rats, intravenous (i.v.) self-administration for the opioid agonist, remifentanil was established during the light cycle. The experiment was conducted within subjects and each session was made up of five (30-minute) components starting under extinction condition and increasing remifentanil doses per component (0.1, 0.32, 1, 3.2 µg/kg/infusion). In these rats trained to self-administer remifentanil, the selectivity of intraperitonially (i.p.) administered test compounds (MG, 7-OH-MG, and MG-P) to antagonize responding for remifentanil vs. a non-opioid, cocaine, was assessed. The abuse potential of MG, 7-OH-MG, and MG-P was also studied within the same cohort. In a separate cohort (4 males and 4 females) of Sprague Dawley rats, rats received escalating doses of morphine (10-40 mg/kg, b.i.d.) paired with either vehicle, or MG (32, 56 mg/kg) over the course of 6 days. On day 6 all rats were given naltrexone (10 mg/kg), along with either their vehicle or MG treatment. Opioid withdrawal was assessed via counting instances of naltrexone-induced diarrhea.
Results: Remifentanil maintained self-administration above extinction levels at most doses tested (0.32, 1, and 3.2 µg/kg/infusion). Each of the kratom alkaloids, MG, 7-OH-MG, and MG-P, were 3- to 4-fold more potent to decrease remifentanil response rates (alkaloid ED50 values: 47.8, 1.99, and 3.36 µg/kg, respectively) than they were to decrease the maximum cocaine cross-administration response rates (alkaloid ED50 values: 135, 7.67, and 10.6 µg/kg, respectively). That is, the antagonistic effects of MG, 7-OH-MG, and MGP were relatively specific for the reinforcing effects of remifentanil over those of cocaine. When the test compounds were substituted for i.v. remifentanil, self-administration responding above extinction was maintained by 7-OH-MG and MGP, but not MG. MG (32 mg/kg) attenuated the development of naltrexone-induced morphine withdrawal signs. Conclusions: Overall, these results in rats suggest low MG abuse potential and support the idea that MG holds therapeutic promise as an opioid dependence medication. However, at high doses above naturally occurring MG metabolism, isolated MG active metabolites may possess abuse potential and warrant consideration for further studies.
Supported by National Institute on Drug Abuse grants DA25267 and DA048353.
Gisela A. Camacho-Hernandez1; Andrea Casiraghi1,2; Deborah Rudin3; Dino Luethi3,4; Amarachi V. Okorom1; Therese C. Ku1, Daryl A. Guthrie1; Valentina Straniero2; Ermanno Valoti 2; Gerhard J. Schütz, 4; Harald H. Sitte, 3; Amy H. Newman1
1Medicinal Chemistry Section, National Institute on Drug Abuse-IRP, Baltimore, MD USA; 2Department of Pharmaceutical Sciences, University of Milan, Milan, Italy; 3Institute of Pharmacology, Medical University of Vienna, Vienna, Austria; 4Institute of Applied Physics, TU Wien, Vienna, Austria.
Monoamine transmission is controlled by dopamine (DA), serotonin (5-HT) and norepinephrine (NE) transporters (DAT, SERT and NET respectively), wherein these transmembrane proteins mediate the re-uptake of their respective neurotransmitters from the extracellular space into the cell. Proper function is necessary to achieve homeostasis as dysregulation of monoamine transporters (MATs) is linked to several neuropsychiatric disorders, including substance use disorder. Fluorescently-tagged ligands have proven useful as pharmacological tools to visualize protein expression, localization and distribution in distinct cell systems. Thus, the previously reported fluorescent cocaine-based analogue JHC1-064, has been extensively used as a tool to study MATs due to its high affinity for all three transporters. Nevertheless, lack of selectivity of this fluorescent probe across MATs contributes to certain staining limitations. This has been overcome in SERT with an S-citalopram-based probe, VK2-83, but remains a challenge for DAT and NET. The design of these fluorescent ligands encompasses three structural features: 1) a high affinity parent ligand, 2) a linker of appropriate length, and 3) a fluorescent dye. In this study, utilizing the NET selective inhibitor nisoxetine and the atypical DAT inhibitor JJC8-087 as parent compounds, we have designed and developed rhodamine and diSulfoCy5-labeled fluorescent ligands with high affinity and preferential binding for either NET or DAT. Specifically, the nisoxetine-based fluorescent ligand demonstrated high affinity (Ki = 43 nM) for NET and selectivity over DAT (Ki = 1540 nM) and SERT (Ki = 785 nM) in a competitive radioligand binding assays utilizing rat brain tissue. Visualization of either human NET or human SERT expressed in HEK293 cells, using confocal microscopy, was achieved by this ligand. Additionally, one of our two leads for DAT has 140 nM binding affinity and is ~20 fold more selective over SERT (Ki = 2.9 μM) and ~120 fold more selective over NET (Ki= 17 μM). These lead molecules are currently being tested for staining of DAT in rat tissue and potentially, they will be used to assess DAT expression differences in neurodegenerative and SUD models.
Imad Damaj
Department of Pharmacology and Toxicology, Virginia Commonwealth University
Nicotine and nicotinic acetylcholine receptors (nAChRs) have been explored for the past three decades as targets for pain control, in particular the α7 nAChRs subtypes. I will present the latest developments on the role of α7 nAChRs in neuropathic pain and the potential of α7 nAChRs ligands as potential analgesics. Our studies and others suggest that selective ligands for α7 nAChRs hold promise in the treatment of chronic pain conditions as they lack many of side effects associated with other nicotinic receptor types.
Dai Lu, Ph.D.
Rangel College of Pharmacy, Texas A&M University
Rangel College of Pharmacy, Texas A&M University
A new class of compounds that can regulate the function of cannabinoid receptors (CBRs) have been identified in 2005. Unlike the traditional orthosteric ligands which compete with the endocannabinoids for receptor sites, the new class of compounds do not compete with endocannabinoids and thus were termed as allosteric modulators (AMs) of cannabinoid receptors. Today, both positive allosteric modulators (PAMs) and negative allosteric modulators (NAM) of CB1 receptor have been identified. Some of these CB1 allosteric modulators have been investigated for therapeutic usefulness in different models of disease such as pain, glaucoma, and neurodegeneration. These CB1 PAMs showed promising therapeutical effects. Translation of CB1 allosteric modulators into clinically relevant candidates holds strong value to discover cannabinoid-based medication for some unmet medical needs. In comparison with CB1 orthosteric ligands, allosteric modulators possess stronger capacity to selectively modulate certain specific signaling pathways via functional selectivity or biased signaling, therefore generating novel pharmacology that is difficult or impossible to achieve through traditional orthosteric CB1 ligands. This provides possibility to separate therapeutic effects from unwanted side effects such as psychoactive activities associated with CB1 orthosteric agonists. However, the discovery of allosteric modulators some significant presents challenges beyond those encountered in orthosteric drug discovery. These include 1) the hits identification via in vitro screening, 2) multiple assays for in vitro profiling, 3) principles for lead optimization, 4) rational design and optimization, 5) allosteric agonists vs ago-PAM, 6) formulation for in vivo characterization and 7) translational need of biomarkers etc. Here these challenges will be discussed in relevance of developing CB1 allosteric modulators.
Kelly K. Wingfield1,2, Kayla T. Richardson1,3, Teodora Misic1, Mia B Rubman1,4, Emily J. Yao1, Jacob A. Beierle1,2, Camron D. Bryant1
1. Laboratory of Addiction Genetics, Department of Pharmacology and Experimental Therapeutics and Psychiatry, Boston University School of Medicine
2. T32 Biomolecluar Pharmacology Training Program, Boston University School of Medicine
3. PREP Scholar Program, Boston University
4. NIH/NIDA Summer Undergraduate Fellowship Program
Concomitant with the opioid epidemic, there has been a rise in pregnant women diagnosed with opioid use disorder and cases of infants born with neonatal opioid withdrawal syndrome (NOWS). NOWS refers to symptoms following cessation of prenatal opioid exposure that comprise low body weight, impaired thermoregulation, and hyperirritability. Multiple factors contribute to NOWS severity; however, the genetic factors are unknown. Our goal is to use mouse models to identify genetic variants that contribute to NOWS severity and translate these findings to humans. As a first step, here, we phenotyped genetically similar inbred substrains of FVB/N (NJ, NCrl, NHsd, and NTac) to identify genetic differences in NOWS model traits, including spontaneous hyperalgesia, hypothermia, locomotor agitation, and hyperirritability. In our opioid exposure regimen, neonatal pups were injected twice daily from postnatal day 1 (P1) to P15 with morphine (10 mg/kg, s.c.) or saline. This early life exposure model represents the approximate third trimester in humans regarding developmental timing and this time period is both necessary and sufficient for inducing NOWS model traits. Importantly, this exposure model avoids maternal exposure that could affect maternal care and it also permits precise control of morphine dosing across pups. We assessed several phenotypes during spontaneous morphine withdrawal on P7 and P14, including ultrasonic vocalizations (USVs) and concomitant locomotor activity, followed by nociceptive testing (hot plate and tail-flick latency assays). Morphine pups of all four substrains displayed lower body weights and hypothermia and showed thermal hyperalgesia (reduced nociceptive latency) on the hot plate and tail withdrawal assays compared to saline pups. Interestingly, on P14, but not P7, morphine pups emitted more USVs than saline pups, consistent with the excessive crying in human infants born with NOWS. In mouse pups, USVs indicate emotional states and are emitted in response to thermal change, distress, and maternal separation. USVs are classified into different syllable types based on frequency changes and duration. We explored the composition of USV syllables to determine if certain syllables were associated with the aversive affective state of opioid withdrawal. We also looked for divergence among FVB substrains in USV composition that would provide evidence for genetic differences in the severity of the affective opioid withdrawal state. We implemented the machine learning software, DeepSqueak, to characterize USV syllables. We created a custom supervised classifier to automatically label different USVs based on previously known syllables. During spontaneous withdrawal on P14, we observed a significant increase in the percentage of “complex 3” syllables compared to saline pups across all four substrains; however, there were notable substrain differences in USV composition. On P15, we administered a maintenance dose of morphine (10 mg/kg) or saline to determine if morphine could relieve withdrawal and thus restore the USV profile to a saline-like profile. Morphine induced a significant reduction only in the proportion of complex 3 syllables emitted by morphine pups toward a saline control-like level, suggesting that complex 3 syllables could be a unique marker for an emotional distress signal associated with the opioid withdrawal state.
Astrocyte Elevated Gene-1 (AEG-1) Deletion Selectively Enhances the Antinociceptive Effects of Morphine
Bryan Mckiver1, Eda Köseli1, Shivani Patel1, Apurva Puli1, Katherine Contreras1, Harrison Elder1, Alyssa White-Presley1, Devanand Sarkar2, and M. Imad Damaj1
1Department of Pharmacology and Toxicology, Virginia Commonwealth University, 2Department of Human and Molecular Genetics, Virginia Commonwealth University
1Department of Pharmacology and Toxicology, Virginia Commonwealth University, 2Department of Human and Molecular Genetics, Virginia Commonwealth University
Background: Opioids, such as morphine, are used clinically to treat acute and chronic pain. Morphine functions by activating endogenous µ opioid receptors (MOR), which are expressed in various neuronal and non-neuronal tissues throughout the body. Prolonged usage of morphine and other opioids can often lead to the development of severe negative side effects such as constipation, tolerance, addiction or opioid use disorder (OUD), the development of withdrawal symptoms when drug is not available, and lethal overdose due to severe respiratory depression. Astrocyte Elevated Gene-1 (AEG-1) is a multifunctional protein that regulates inflammation, myeloid immune cell activity and lipid metabolism. Recent studies have shown that there is a significant amount of “cross-talk” between the endogenous opioid system and the immune-mediated inflammatory response, particularly in tissues involved in pain sensory neurotransmission, which could suggest a potential role for AEG-1 in the opioid signaling cascade. Therefore, the goal of this study is to investigate the potential role of AEG-1 in acute and chronic morphine-induced pharmacological effects.
Methods: Adult AEG-1 global knockout (KO) and wild-type (WT) male and female mice (C57BL/6J background) were utilized to assess acute morphine-induced thermal antinociception (tail immersion), hyperlocomotion (locomotor boxes), gastrointestinal (GI) transit inhibition (charcoal transit assay), and respiratory depression (whole-body plethysmography). Chronic morphine studies focused on the effects of AEG-1 deletion on morphine tolerance (tail immersion), and withdrawal behaviors (somatic sign observation). Additionally, we injected mice with morphine and collected plasma samples for mass spectrometry analysis of morphine and morphine-3 glucuronide (M3G) serum levels. Lastly, we assessed acute thermal antinociception using other opioid compounds (oxycodone and buprenorphine) and a synthetic cannabinoid (CP55 940).
Results: AEG-1 KO mice displayed enhanced thermal antinociception following acute and cumulative dosing with morphine compared to their WT counterparts. Pretreatment with low dose naloxone, an opioid receptor antagonist, blocked the enhancement of morphine thermal antinociception in AEG-1 KO mice. No significant differences in morphine-induced hyperlocomotion, GI transit inhibition, or respiratory depression were observed between AEG-1 KO and WT mice. Chronic morphine treated AEG-1 KO mice displayed reductions in tolerance development and total somatic signs during precipitated withdrawal compared to their WT counterparts. Similar to morphine, AEG-1 KO mice treated with oxycodone and buprenorphine, separately, displayed enhanced thermal antinociception compared to their WT counterparts. However, administration of CP55 940 produced no significant difference in antinociceptive effect between AEG-1 KO and WT mice.
Conclusions: Our data suggest that AEG-1 deletion enhances the antinociceptive effects of morphine, and other MOR agonist, reduces tolerance to chronic morphine treatment and reduces the symptoms of morphine withdrawal. However, AEG-1 deletion does not impact morphine-induced locomotor activity, GI transit inhibition, or respiratory depression. Lastly, this effect appears to be specific to opioid receptor mediated antinociception. Overall, our results suggest that AEG-1 may function as an endogenous modulator of opioid-induced antinociception.
Methods: Adult AEG-1 global knockout (KO) and wild-type (WT) male and female mice (C57BL/6J background) were utilized to assess acute morphine-induced thermal antinociception (tail immersion), hyperlocomotion (locomotor boxes), gastrointestinal (GI) transit inhibition (charcoal transit assay), and respiratory depression (whole-body plethysmography). Chronic morphine studies focused on the effects of AEG-1 deletion on morphine tolerance (tail immersion), and withdrawal behaviors (somatic sign observation). Additionally, we injected mice with morphine and collected plasma samples for mass spectrometry analysis of morphine and morphine-3 glucuronide (M3G) serum levels. Lastly, we assessed acute thermal antinociception using other opioid compounds (oxycodone and buprenorphine) and a synthetic cannabinoid (CP55 940).
Results: AEG-1 KO mice displayed enhanced thermal antinociception following acute and cumulative dosing with morphine compared to their WT counterparts. Pretreatment with low dose naloxone, an opioid receptor antagonist, blocked the enhancement of morphine thermal antinociception in AEG-1 KO mice. No significant differences in morphine-induced hyperlocomotion, GI transit inhibition, or respiratory depression were observed between AEG-1 KO and WT mice. Chronic morphine treated AEG-1 KO mice displayed reductions in tolerance development and total somatic signs during precipitated withdrawal compared to their WT counterparts. Similar to morphine, AEG-1 KO mice treated with oxycodone and buprenorphine, separately, displayed enhanced thermal antinociception compared to their WT counterparts. However, administration of CP55 940 produced no significant difference in antinociceptive effect between AEG-1 KO and WT mice.
Conclusions: Our data suggest that AEG-1 deletion enhances the antinociceptive effects of morphine, and other MOR agonist, reduces tolerance to chronic morphine treatment and reduces the symptoms of morphine withdrawal. However, AEG-1 deletion does not impact morphine-induced locomotor activity, GI transit inhibition, or respiratory depression. Lastly, this effect appears to be specific to opioid receptor mediated antinociception. Overall, our results suggest that AEG-1 may function as an endogenous modulator of opioid-induced antinociception.